日本語 / English
特別推進研究
研究計画
平成22年度
(1)配位子に対する置換基有効ポテンシャル法の開発:
官能基、置換基の電子的効果を取り込む有効ポテンシャル法を開発する。これまではσ電子系であるアルキルホスフィン配位子の孤立電子対軌道のモデル化を行ってきた。ホスフィンは多くの錯体で使用されているが、アルキルホスフィンと共に、トリフェニルホスフィンが極めて多くの錯体に使用されている。ノーベル賞となったWillkinson錯体もトリフェニルホスフィンを使用している。また、ペンタメチルシクロペンタジエニル配位子も非常に多くの錯体で使用されている。しかし、これらの配位子は分子サイズが大きいため、高精度のCCSD(T)やMP4計算は不可能である。本研究ではこれらの錯体の構造、反応過程の理論・計算化学研究を高精度に行うため、有効ポテンシャル法をこれらのπ電子系を持つ配位子に拡張する。
開発したポテンシャルが正しい結果を与えるか、代表的なホスフィン錯体やペンタメチルシクロペンタジエニル錯体に適用し、エネルギー変化が化学的精度(誤差1 kcal/mol)未満であるか、を確認する。
(2) 高精度電子状態理論のハイブリッド化:
励起状態を高精度で記述することの可能な代表的なプ計算法はSAC-CI法である。しかし、SAC-CI法は計算コストが高く、大規模系には応用できない。とりわけ、遷移金属錯体では精度が有機分子に比べて劣っている。General-R法のようにSAC-CI法に高次の励起演算子を考慮する方法を開発する。General-R法は高精度であるが、非常にコストが高く、数原子分子しか適用できない。本方法では中心部分に高次項を考慮したSAC-CI法、配位子にFOC-EP法、周辺部分はFMOで取り込むハイブリッド化計算法を開発する。この方法をGAMESS, Gaussian03などの汎用プログラムに組み込む。
(3)複雑な電子状態を有する多核金属錯体の構造と結合性、電子状態の解明: 金属多重結合を持つ多核錯体の構造と電子状態の解明:
現代の電子状態理論をもってしても、金属多重結合を持つ多核金属錯体の高精度計算を、化学的精度で行うことは未だ不可能である。本研究では、配位子の電子的効果をFOC-EP法で取り込み、中心部分にCAS-PT2法、もしくは、RAS-PT2法を、周辺部分はFMO的にMP2法、もしくはDFT法で取り込み、金属多核錯体の高精度計算を行なう。結合性の検討は、FragementのNatural Orbitalで全系のNatural Orbitalを展開し、occupation数を考慮して、σ-, π-, δ-結合性の寄与の大小評価し、金属多重結合とd軌道の広がり、d, s, p軌道エネルギーとの関連を明らかにする。この解析により、第一、第二、第三遷移周期元素の多重結合の特徴を遷移金属元素の性質(d軌道の広がりとd電子数、d, s, p軌道エネルギ-差)から解明する。研究対象としては一連のCr(0), Cr(I), Cr(II)二核錯体、類似のMo錯体を取り上げ、Cr-Cr間結合が異常に短い理由を明らかにする。
(4) 遷移金属元素と高周期元素の単結合および多重結合の理論的研究:
遷移金属とシリル、ゲルミルなどとの単結合、シリレン、ゲルミレンなどの二重結合、シリリン、ゲルミリンなどの三重結合の理論的研究をCAS-PT2法、もしくは、RAS-PT2法で行い、電子相関効果が単結合と多重結合でどのようにことなるのか、静的相関が大きいのか、小さいのか、実質的な結合次数はどの程度なのか、などの結合論の基本的な知見を明らかにする。置換基の電子的効果はFOC-EP法で、また、立体効果はDFT法で取り込む。(3)と同様に、結合性はFragementのNatural Orbitalで全系のNatural Orbitalを展開し、occupation数の解析と合わせて、結合性の寄与の大小評価、d軌道の広がり、エネルギー準位との関連を明らかにする。対象としては、M-Si単結合化学種として、R3Si-M(Cl)(PR3)2 (M=Pt or Pd)、二重結合化学種として、R2Si=Cr(CO)5および類似Mo錯体、三重結合化学種として、RSi≡Mo(NR)3を取り上げる。
(5) 遷移金属錯体による触媒反応の理論的研究と予測:
初年度は、有機合成分野で重要な反応となっているクロスカップリング反応およびランタンなどのランタナイド金属による官能基導入エチレン重合反応を取り上げる。クロスカップリング反応にはパラジウムが触媒として使用される場合がほとんどである。しかし、最近はロジウムも使用される。ロジウムによるクロスカップリング反応の反応機構を解明し、各素反応過程をパラジウム錯体のそれと比較する。それらによりロジウムとパラジウムの触媒としての特徴を明らかにする。構造はDFT法で最適化するが、エネルギー変化はFOC-EPを使用したCCSD(T)法で求める。遷移状態や重要な中間体の相互作用は、全系の分子軌道をFragmentの分子軌道の一次結合であらわすことにより解析する。
ランタナイド錯体によるエチレン重合反応とアミノ基導入反応の触媒サイクルについて理論計算を行い、律速過程を明らかにする。エネルギー変化は上述したようにFOC-EP法を用いたCCSD(T)法で求める。
これらの計算結果を得た後、配位子の電子的効果をFOC-EP法により系統的に変化させ、最適な配位子の電子的性質を理論予測する。その後、実際に存在する配位子を探索し、その遷移金属錯体について触媒反応全体の構造、エネルギー変化を求め、理論予測を行なう。
平成23年度以降
(1) 金属表面用有効ポテンシャル法の開発:
金属表面に吸着した化学種の電子状態計算は周期境界条件を考慮したスラブモデルで行われる例が多い。この場合、金属表面を切り出すという不自然なモデル化は無いから、一見、正しいモデルのように感じられる。しかし、平面波基底しか用いられない。吸着相互作用は局所的である場合が多く、また、吸着種の電子状態は局所的であるから、平面波基底がそれらの記述に適しているとは思われない。また、SAC-CI法やCCSD(T)法などの高精度post Hartree-Fock法はコストの点から適用できない。本研究ではSAC-CI法やCCSD(T)法が適用可能なクラスターモデルを採用し、周辺効果はFOC-EP法で取り込むことを考える。そのモデル化として、パラジウムや白金についてパラメーターを開発し、もっとも一般的なCOの吸着エネルギーを実験結果と比較し、この方法の妥当性を検討する。
(2)金属表面吸着種の理論化学研究:
パラジウムや白金表面をクラスターモデル化し、エチレン、COなどの基本的な分子が吸着した構造をDFT法で求め、SAC-CI法で励起エネルギーを求め、帰属を行なう。
また、酸素や水素の解離吸着のエネルギー変化をCCSD(T)法で求め、吸着過程の高精度理解を達成する。
(3) 高周期へテロ元素の多重結合を含む遷移金属複合系の構造、結合性、電子状態の解明:
実験化学、理論化学双方で興味深い研究対象となっているケイ素やゲルマニウムなどの多重結合の電子状態、結合性を取り上げ、それらの遷移金属錯体の結合性を解明することを目的とする。これまで、これらの多重結合の理論的研究は永瀬らにより精力的に進められてきた。しかし、π-アリル、π-プロパルギル、シクロブタジエニルのケイ素置換体などの共役化合物の電子状態、結合性は、ほとんど検討されていない。ここでは、これらの化学種の結合性と電子状態を明らかとし、炭素化学種との比較を通して、特徴を明らかにする。さらにそれらの遷移金属錯体を取り上げ、配位結合が炭素類似体とどのように異なるか、どう特徴つけられるのか、を明らかにする。方法としてはDFT法でなく、CCSD(T)法、あるいは多配置性が大きい場合はCASPT2法を用い、置換基の電子的効果はFOC-EP法で検討する。
具体的には、最近、関口らにより合成された、以下の化学種を取り上げる。いずれも芳香族性の有無、金属との非古典的な相互作用が予想される点で分子科学的に興味深い化合物である。
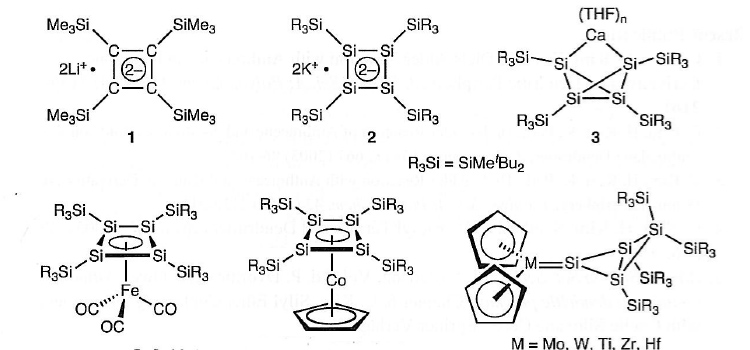
これらの化学種のSi-Si間結合、M-Si間結合がどのように炭素のそれと異なるか、類似のGeのそれと異なるのか、明らかにする。また、新しい金属錯体化学種の理論予測を行い、これらの新しい結合を持つと期待される化合物の単離に必要な条件を実験分野に提供する。
(4) 高周期へテロ元素や遷移金属元素を含むナノカーボン化合物の構造と機能の理論的研究:
フラーレンに金属を内包した金属内包フラーレンは通常のフラーレンに比べて、電子状態が大きく異なる。その結果、通常のフラーレンと異なる反応性を示す。機能については、不明な点がほとんどである。しかし、HOMOエネルギーが高くなることから電子移動が容易になると期待されるし、吸収スペクトルや発光スペクトルが大きく変化すると期待される。本研究では金属内包フラーレン、ヘテロ元素導入金属内包フラーレンを取り上げ、その構造、電子状態、安定性、反応性を理論計算から明らかにし、これらの新しいナノカーボン化合物の基本的性質を解明する。
カーボンナノチューブについては、高周期へテロ元素の面への導入、エッジへの導入により、どのように電子状態や機能が変化するか、を解明する。
また、遷移金属錯体はフラーレンやカーボンナノチューブと錯体を形成する。これらの錯体は理論計算の対象となっているが、榊らの研究を除いて、DFT法を無批判に応用したものばかりである。しかし、榊らの理論的研究ではDFT法はこれらの遷移金属作体の結合エネルギーを過小評価することが明らかにされている。本研究では、安易なDFT法の利用をせず、ハイブリッド化した高精度理論計算法を適用し、重要部分についてはpost Hartree-Fock法で検討する。
(5)遷移金属錯体による触媒反応の理論的研究と予測:
平成22年度に引き続き、錯体触媒反応の反応機構の解明と新規触媒の予測を行なう。
(5-1) ルテニウム、ロジウム錯体触媒による二酸化炭素水素化化反応:
触媒サイクルを理論的に解明し、中心金属の触媒作用メカニズムを分子論的に明らかにし、代替金属の可能性を予測する。代替金属としては、水素化触媒作用があり、二酸化炭素と相互作用が可能なFeを取り上げる。
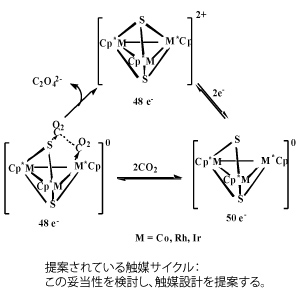
(5-2) ロジウム錯体触媒による二酸化炭素固定化反応:
田中らにより報告されたロジウム錯体による二酸化炭素からシュウ酸への変換反応を取り上げる。中間体が不明であるので、中間体をまずもとめ、次に、遷移状態を求め、最終的に触媒サイクルを理論的に解明する。中心金属をロジウムからコバルトなどに変換可能か、その触媒作用メカニズムを分子論的に明らかにし、それに基づいて検討する。
(5-3)熊田-玉尾クロスカップリング反応:
多くのクロスカップリング反応がパラジウムを触媒に使用する中で、熊田-玉尾クロスカップリング反応はニッケルを触媒とする。従って、ニッケルの触媒作用を明らかにすることが出来れば、パラジウムからニッケルへの変換に役立てることが出来ると考える。特に、ラジカル機構と協奏機構の比較を行なう必要がある。従って、電子状態理論としてはCCSD(T)法もしくはCASPT2法を使用することは不可欠である。我々が開発したFOC-EP法でこれらのコストの高い方法が利用可能となる。
(5-4)園頭クロスカップリング反応:
この反応は共役C-C結合を形成するために有力である。パラジウムと有機銅化合物の組み合わせで進行する。有機銅の役割、特に、トランスメタル化でのふるまいを解明し、有軌銅以外にどのようなカップリングパートナーが使用可能か、理論的に予測する。
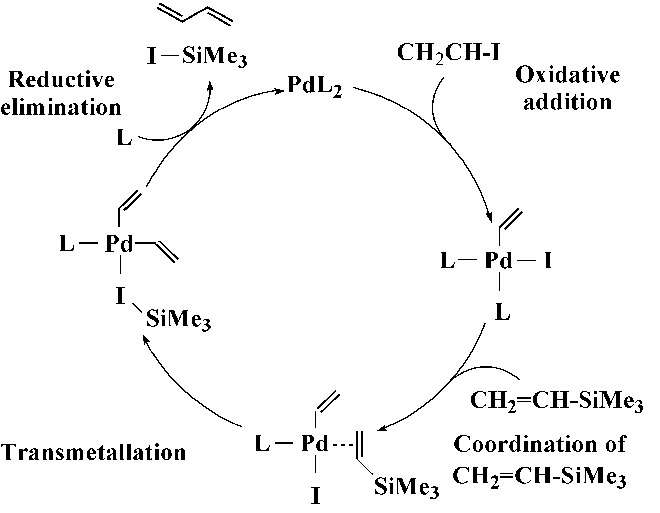
クロスカップリングの一般的な触媒サイクル:これをNi錯体を触媒とする熊田-玉尾カップリングと比較。PdとNiの比較を通して新規触媒の提案を試みる。
(5-5)酸化反応触媒の理論的予測:
酸化反応は制御が困難な代表的な反応である。我々はすでに遷移金属による酸素分子の活性化過程の詳細な解明に成功している。得られた酸素錯体の反応性を各種基質に対して検討する。特に、炭化水素の酸化反応を取り上げ、カテコールジオキシゲナーゼモデル鉄錯体のよる芳香族炭化水素の酸化反応、エチレンからエチレンオキシドへの変化過程を検討する。前者は既にSiegbahnらにより検討されているが、実験化学者によると反応機構に納得できない点があると言われている。反応機構を再検討するとともに、類似の金属錯体による同様の酸化反応を取り上げ、新規触媒の理論設計を試みる。
後者の反応は銀表面反応であるが、これまで多数の理論的研究が行なわれているが、いずれもクラスターモデルである。我々は、金属表面効果をFOC-EPで取り込み、高精度の表面反応の理論的研究を行うことが可能である。この研究により銀触媒によるエポキシ化の触媒反応過程を検討しなおし、それに基づいて、新しい触媒探索を試みる。